
Spring/Summer 2021
AI Uses Language Rules to Simulate Molecular Motions on Bridges
Recreates Known Chemical Rules, Opens Door to Improved Vaccines, Drugs, Industrial Processes
by Ken Chiacchia
Better predictions of molecular motions could lead to improved vaccines, drugs and any number of improved industrial chemical processes. A team from the University of Maryland used natural language processing artificial intelligence (AI) on PSC’s Bridges platform to re-create known chemistry, showing that AI may be able to reduce molecular dynamics to rules of grammar and syntax. The work offers the potential for leaping ahead of current computational limits in the field.
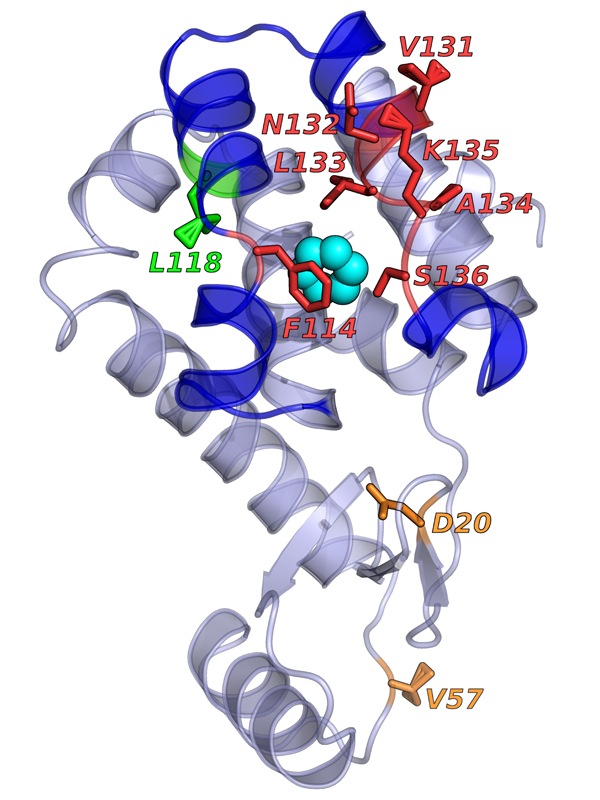
The AI-based method successfully predicted which amino acids (in red, labeled with their symbol letters and their number in the protein’s amino-acid chain) would be critical for the binding of benzene (blue circles) to the lysozyme protein. From Wang Y et al. Past–future information bottleneck for sampling molecular reaction coordinate simultaneously with thermodynamics and kinetics. Nature Communications (2019) 10:3573.

Pratyush Tiwary, University of Maryland
Why It’s Important
The movements of molecules—molecular dynamics—loom large for our health, safety and economy. Our ability to fight COVID-19 relies on our antibodies’ ability to wrap around virus proteins. Cleanup of polluted environments can be improved by engineering microbes to eat the bad stuff. Refinery chemical processes can produce cleaner and more efficient or dirtier and more wasteful fuels. Better predictions of the chemistry could make us safer, cleaner, wealthier.
Modern supercomputers have revolutionized our ability to simulate molecular dynamics. These predictions direct laboratory experiments to create better vaccines, drugs and other chemical reactions. But the limits of even powerful computers still restrict the field.
“Natural language prediction is not an easy problem. You can’t just look at the words. These algorithms, they’re quite fancy. They tend to take into account the context of the words: what is the bigger picture? So the question is, can we take a molecular dynamics trajectory and … map it into an abstract language? … Maybe we can use language processing tools … to learn a better language ‘spoken’ by these molecules.”—Pratyush Tiwary, University of Maryland
Pratyush Tiwary and his students at the University of Maryland wondered whether they could completely change up scientists’ approach to molecular dynamics. Would it be possible to harness the power of AI to detect a simpler set of rules? They turned to natural language processing, a set of so-called recurrent neural network tools loosely based on the workings of living brains. It’s the technology that powers word suggestions on smartphones. Could natural language processing suggest next molecular movements in the same way?
They would need access to a powerful supercomputer to make the approach work. They turned to PSC’s Bridges platform.
How PSC Helped
Natural language processing works by understanding what’s been said already as well as what’s likely to be said next. The AI does this by learning. You begin with a set of sentences in which each word is labeled to explain its meaning and role in the sentence. The computer program processes what’s already been “said” via a series of layers, each representing a different concept in language structure, with many connections between the nodes in each layer. The output of these layers tries to predict the next word. The AI removes connections when it’s wrong and tries again, removing and reconnecting connections until it’s predicting correctly.
After this training step, the scientists present the AI with an unlabeled testing data set. The AI is scored on its ability to predict next words. Like a student in school, the AI goes back and forth between training and testing until its performance is good enough to try to solve a real problem with new data.
Tiwary and his team—graduate students Yihang Wang, Sun-Ting Tsai, Zachary Smith and others— tested their AI-based approaches by moving their computations between the then state-of-the-art NVIDIA Tesla P100 GPU nodes of Bridges and the platform’s CPU nodes. The AI used the GPUs to learn and predict; the CPUs performed molecular dynamics to test the predictions. Such movement can be a tricky process on other systems, but is simplified by software developed at PSC.
The team trained and tested their AI in a number of trial systems. Among these, they simulated the twists of two critical chemical bonds in the simple molecule alanine dipeptide. They duplicated the binding of a benzene molecule to the protein lysozyme. And they recreated the workings of a riboswitch, a molecular switch that changes which amino acid is inserted into a protein chain as it’s being assembled by a living cell.
“We used a lot of Bridges CPU and Bridges GPU [nodes]. We used the P100s heavily. And they are very fast for this. The classical molecular dynamics is run on the CPUs. Bridges has such nice installations of [the software], and scaling is quite efficient … We were doing molecular dynamics on CPUs for an extended amount of time, then coming back to the GPUs to train the AI model, then going back to the CPUs.”—Pratyush Tiwary, University of Maryland
The team’s AI made excellent predictions in all of these trial systems, providing a crucial proof of concept for the method. But it did more. Examining the text-prediction algorithm created by the AI, the scientists realized that the machine, with no prompting from them, had recreated path entropy, a concept introduced by pioneering scientists like Boltzmann, Shannon and Jaynes. A rule for how transfers of energy restrict physical processes, entropy is a cornerstone of modern physics and chemistry. This gave the researchers confidence that their AI is fundamentally on the right track. They published their results in two papers in the journal Nature Communications, in August 2019 and October 2020.
The Maryland scientists plan to expand their work to more complicated systems, possibly using the more-advanced V100 GPUs in the Bridges-AI system and Bridges’ replacement, the greatly expanded Bridges-2 platform. Their hope is to simplify the task of molecular dynamics predictions to leap ahead of the current limitations of the most powerful computing systems, leading to improved medical and industrial tools.